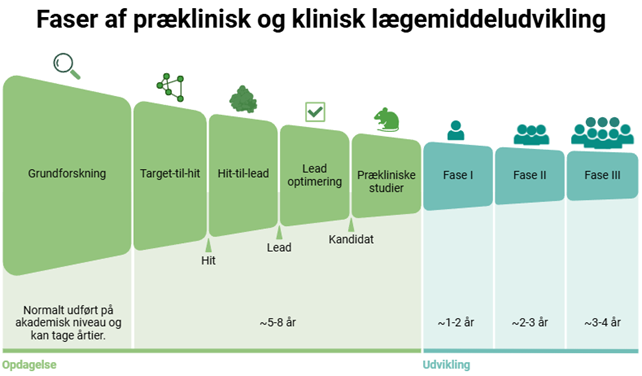
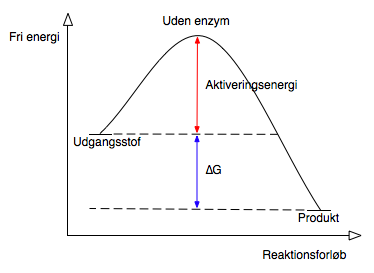
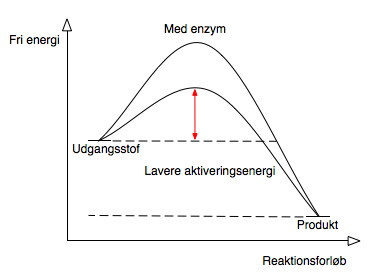
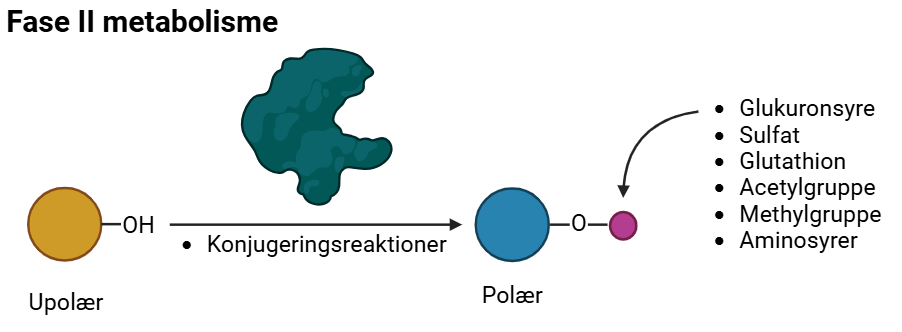
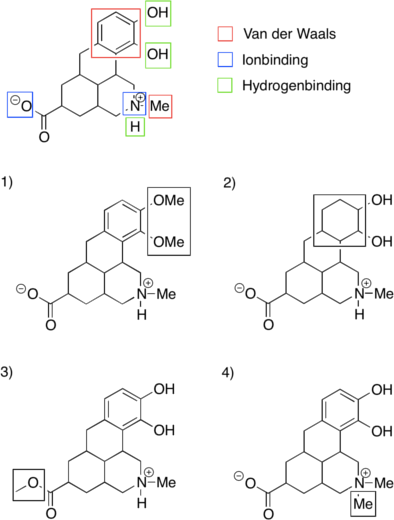
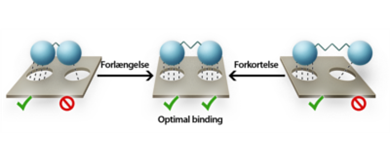
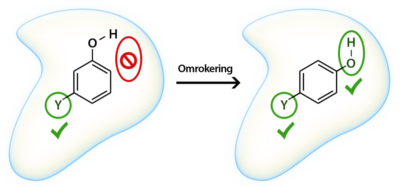
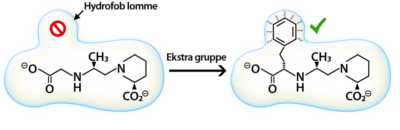
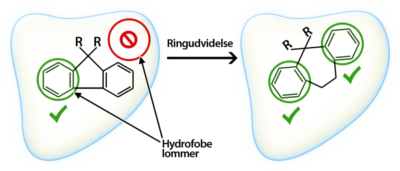
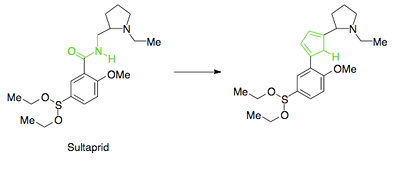
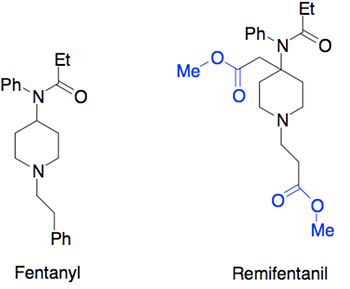
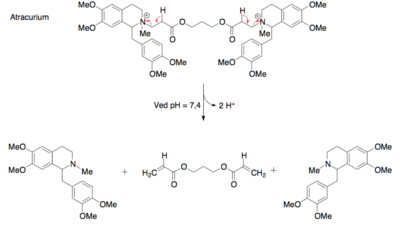
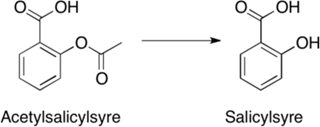
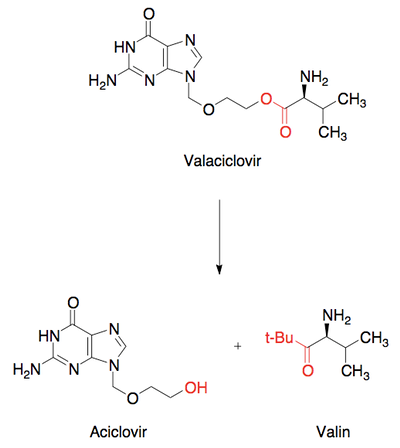
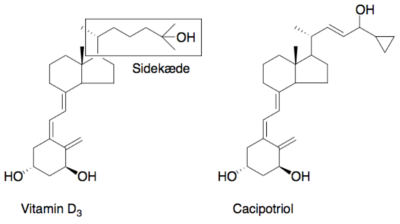
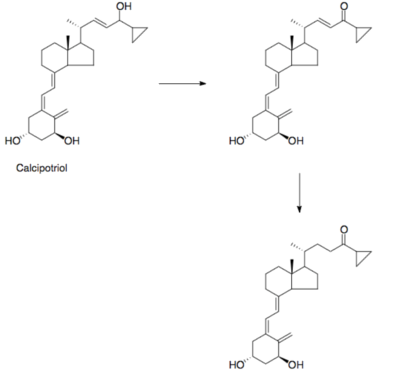
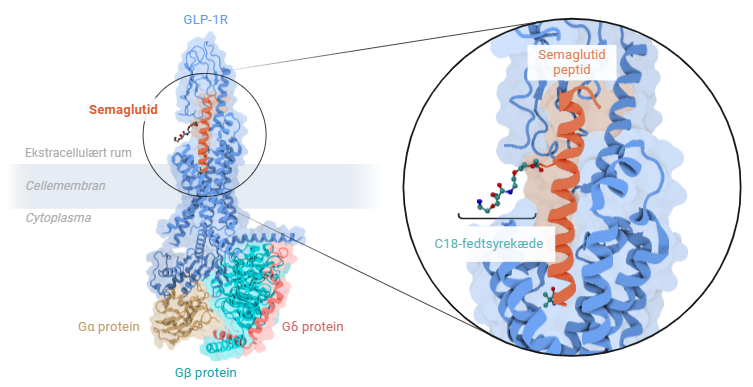
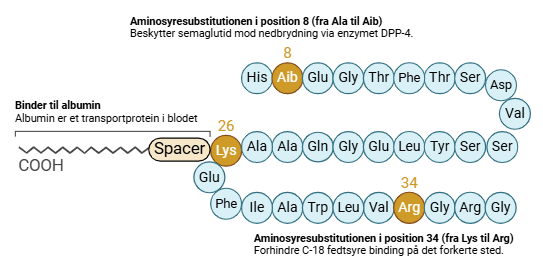
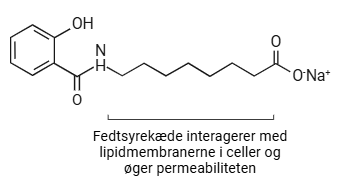
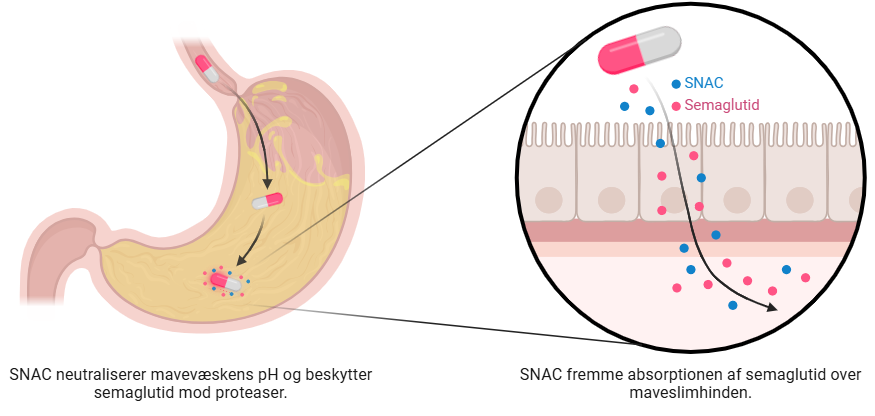
Når man skal udvikle et nyt lægemiddel, starter det hele med grundforskning. Dette er også idéfasen, hvor den første udgave af lægemidlet enten bliver udvundet (fundet i naturen) eller syntetiseret (i et laboratorie). Ofte er formålet at finde en løsning på, hvordan vi kan hjælpe kroppen med at bekæmpe en kendt sygdom eller bivirkninger, der forstyrrer vores dagligdag. Det er essentielt at udvælge det rigtige target, eftersom lægemidlet skal designes efter dette target.
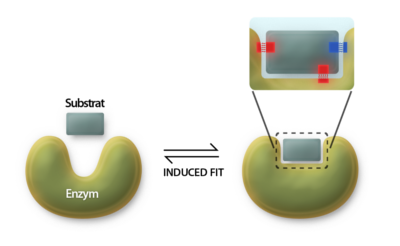
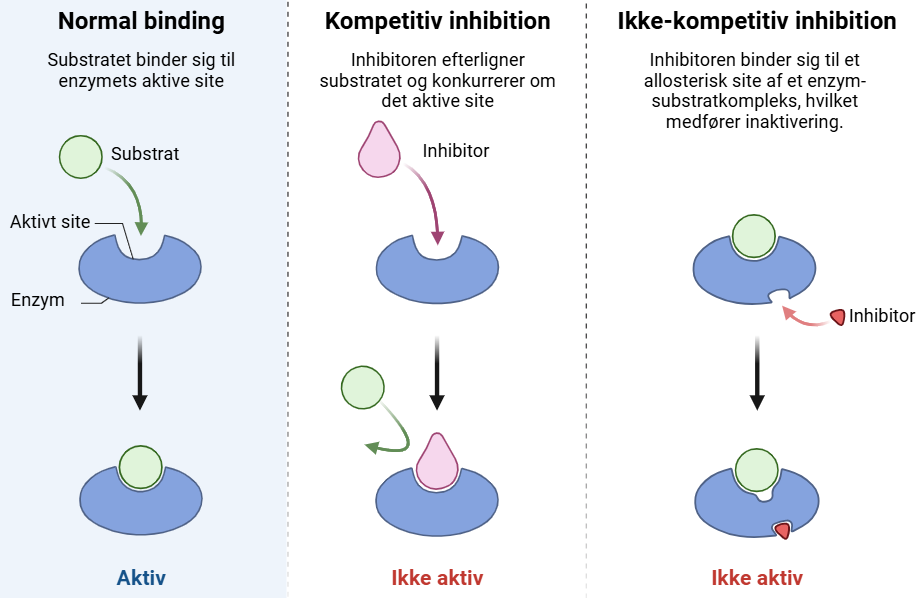
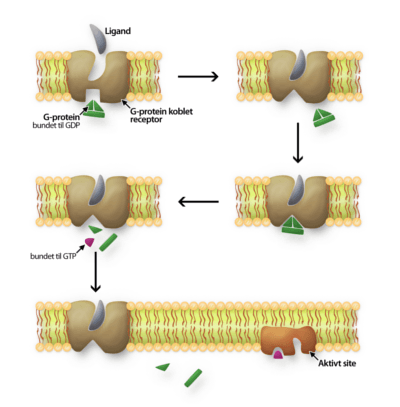
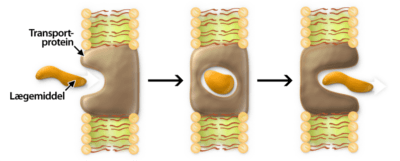
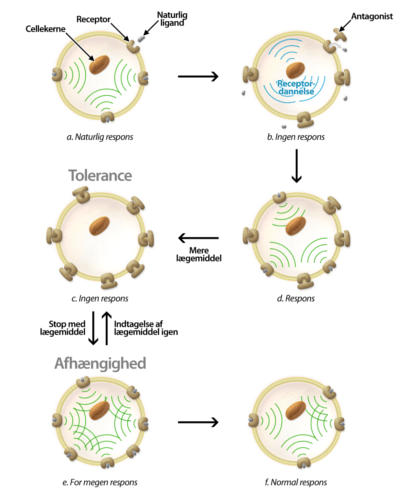
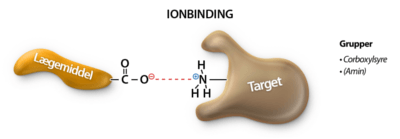
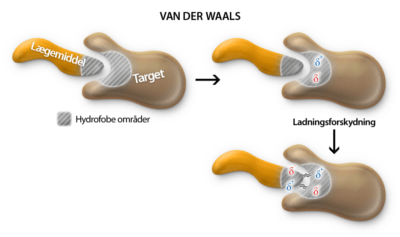
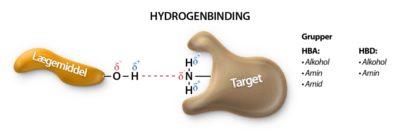
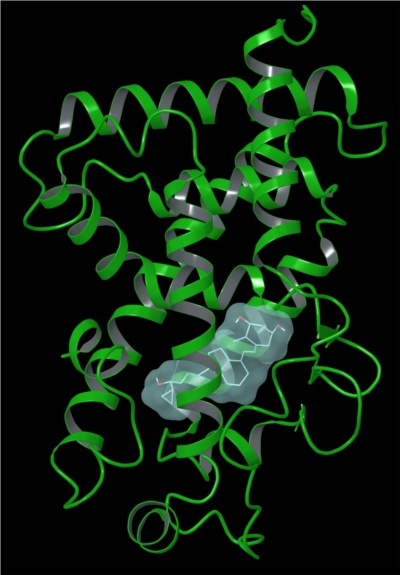
Et target er det sted i kroppen, hvor et lægemiddel skal binde sig for at give den ønskede effekt. Et target kan være mange ting, men er stort set altid et eller flere proteiner i kroppen. Dette kan du læse mere om under den næste underside: “Proteiner som drug targets”.
Når vi har fundet et target, kan vi udvikle de første versioner af vores lægemiddel. Disse er ofte kendt som hits. Når vi arbejder videre i laboratoriet, kan hits videreudvikles til leads og til sidst til lægemiddelkandidater. Når man udvikler ny medicin i laboratoriet, starter man med in vitro-eksperimenter. Dette er en bred betegnelse for eksperimenter udført uden en levende organisme. Hvis en lægemiddelkandidat viser lovende resultater, er næste step in vivo-eksperimenter. In vivo er en betegnelse for eksperimenter udført på dyr. Valget af dyreart afhænger af, hvad man ønsker at undersøge. Dyrenes størrelse påvirker også omkostningerne ved forsøget samt kravene til de nødvendige tilladelser.
Efter det, der samlet set kaldes prækliniske studier, kan et lægemiddel testes yderligere via kliniske studier. Det er her, vi for første gang afprøver lægemidlet på mennesker. De kliniske studier opdeles overordnet i fire faser, hvoraf de første tre udføres inden godkendelse. Fase IV finder sted efter godkendelse og markedsføring, da medicinalvirksomheden er forpligtet til at overvåge langtidsbivirkninger.