
Udvikling af nye lægemidler
Introduktion
Brugen af medicin til behandling af sygdomme har været udbredt, næsten lige så længe som mennesket har eksisteret. Helt tilbage til 400 år f.Kr. benyttede den græske naturlæge Hippokrates ekstrakt udvundet fra piletræets bark til behandling af smerter. Det aktive stof i barken blev senere videreudviklet til acetylsalicylsyre, som er det aktive stof i bl.a. Ipren og Aspirin. Acetylsalicylsyre er i dag et af de mest anvendte smertestillende lægemidler i verden.

Figur 1. Strukturformler for salicylsyre og det videreudviklede acetylsalicylsyre.
Et lægemiddel er et biologisk aktivt stof, som kan bruges til behandling af en sygdom eller bestemte symptomer. Lægemidler binder til biologiske targets i kroppen. Disse targets kan f.eks. kan være proteiner, sakkarider, nukleinsyrer eller lipider. Gode lægemidler virker specifikt og har derfor få bivirkninger. Lægemidler er som regel organiske molekyler, hvis strukturelle opbygning og sidegrupper har stor betydning for deres binding og effekt i kroppen.
At udvikle lægemidler er en stor udfordring, som ikke bliver lettere, når lægemidlet skal virke i hjernen. Hjernen er opbygget af komplekse neurale netværk, som benytter mange forskellige kemiske transmittersystemer, hvilket gør det kompliceret at forstå årsagen til sygdomme i hjernen. Hjernen er samtidigt svær at ramme, da den besidder en naturlig, selektiv barriere, som bestemmer, hvad der kommer ind og ud. Der går ca. 10 til 15 år, fra man får ideen til et lægemiddel, og til det er færdigudviklet. At være drughunter er en udfordrende opgave, som kræver megen viden om lægemidler, og om hvordan de virker.
Når nye stoffer opdages
Tidligere i historien blev de fleste lægemiddelkandidater fundet tilfældigt. Et eksempel var, da Alexander Fleming i 1928 fandt ud af, at svampen Penicillium notatum producerede et stof, der effektivt slog bakterier ihjel. Penicillin revolutionerede måden, hvorpå man behandlede infektioner. Udviklingen af penicillin er bare et af mange eksempler på, hvordan svampe, planter, bakterier og dyr danner biologisk aktive stoffer, der kan bruges som lægemidler. I dag benytter de fleste medicinalvirksomheder, som bl.a. Lundbeck, sig af samme metode, dog på lidt anderledes vis.

Figur 2. Alexander Fleming (1881-1955). Strukturformel for penicillin.
Nye lægemiddelkandidater findes i dag ved at bruge store robotter, som screener kemiske biblioteker efter ”trial and error”-princippet. Ved en screening undersøges stofferne i de kemiske biblioteker for deres medicinske effekt på forskellige cellekulturer. Bibliotekerne kan både være tilfældigt syntetiseret med kombinatorisk kemi, eller kan være stoffer oprenset fra naturen. Som beskrevet tidligere er naturen rig på stoffer med biologisk aktivitet. Ved at screene kan mange stoffer hurtigt undersøges for deres bindingsegenskaber til biologiske targets. Nye potentielle lægemidler med de ønskede farmakologiske virkninger identificeres som hits. En enkelt screening giver tit flere hits, men der er stadig et stykke vej til et endeligt lægemiddel.
I dag laves der ofte en tredimensionel model, af den receptor eller det enzym, man ønsker at ramme med et lægemiddel. Dette kunne f.eks. være NMDA-receptorerne i hjernen. Disse receptorer har central betydning, når man behandler Alzheimers sygdom (se dropdown nedenfor). Ved hjælp af røntgenstrukturanalyse og kemiprogrammer til tredimensionel analyse kan forskerne modellere organiske molekyler på baggrund af viden om targets tredimensionelle struktur. Modellen kan bruges til at afprøve lægemiddelkandidater på target. Disse undersøgelser omtales ofte som in silico tests.

NMDA-receptoren har stor betydning for den excitatoriske neurotransmission i hjernen, hvilket betyder, at den er med til at forstærke signaloverførsel mellem nervecellerne i hjernen. Receptoren findes i nervesynapser, hvor den aktiveres, når glutamat bindes til den. Receptoren aktiveres af både glutamat og aspartat, men aspartat virker dog ikke lige så kraftigt som glutamat. Receptoren kræver desuden en co-agonist som glycin for at sikre en optimal åbning af receptorens ionkanaler. Glutamat er det primære excitatoriske neurotransmitterstof, som bruges i alle områder af hjernen. NMDA-receptoren er permeabel for Ca2+ ioner (se figur 6). Ca2+ strømningen igennem NMDA-receptorens ionkanal gør den helt speciel, da Ca2+ kan igangsætte mange forskellige processer i cellen. Forskere mener bl.a., at indstrømningen af Ca2+ er skyld i de molekylære ændringer, der har betydning for lagringen af langtidshukommelsen.

Figur 4. Strukturformler for aminosyrerne glutamat, aspartat og glycin.
Når enten glutamat eller aspartat binder til NMDA-receptoren, vil ionkanalen åbnes, og Ca2+ og Na+ strømmer ind i cellen, mens K+ strømmer ud. Ionstrømmene fører til en depolarisering af cellens membran, som ved tilstrækkelig stimulering kan skabe en nerveimpuls i neuronet. NMDA-receptoren er et primært biologisk target for stoffet memantin, som bruges til behandling af Alzheimers sygdom.

Figur 5. Ved aktivering åbnes NMDA-receptorens ionkanal, hvorefter calciumioner og natriumioner strømmer ind i cellen, mens kaliumionerne strømmer ud. Ionstrømmen medfører en depolarisering af cellens membran.
Hvordan virker lægemidler?
De fleste lægemidler påvirker helt bestemte biokemiske processer, som har indflydelse på, hvordan kroppens celler fungerer. Man designer ofte lægemidler med det formål, at den normale biologiske funktion påvirkes. Enten kan funktionen af det biologiske target nedsættes eller forbedres. Stoffer, der binder til en receptor og aktiverer receptoren, kaldes agonister (forbedre funktionen), mens stoffer, der hæmmer aktiveringen af receptoren, kaldes antagonister (nedsætter funktionen) (se figur 6). De fleste lægemidler virker som enten agonister eller antagonister.

Figur 6. Til venstre ses virkningen af en agonist, og til højre ses virkningen af en antagonist.
Da lægemidler tit ligner kroppens egne signalstoffer, skal de ofte konkurrere med disse om at binde deres mål (kaldet target). Hvis target er et enzym, kan lægemidlet f.eks. binde til enzymets regulatoriske center eller enzymets aktive center. Binding til det aktive center påvirker enzymet direkte, fordi det er her, de naturlige signalstoffer eller substrater binder og gennemgår den kemiske reaktion. Binding til det regulatoriske center kan derimod forårsage en ændring i enzymets tredimensionelle struktur (konformation), som indirekte ændrer formen af det aktive center. Det kan gøre, at signalstoffer eller substrater enten ikke kan binde sig, eller at de binder sig lettere end normalt. Hvis target er en receptor, vil lægemidlet ofte blokere de bestemte dele af receptoren, som binder signalmolekyler eller på anden måde har central betydning for receptorens funktion (se om NMDA- receptoren).

Figur 7. Enzym med aktivt og regulatorisk center.
For at forstå, hvordan et lægemiddel binder til et enzym eller en receptor, er det vigtigt at kende til intermolekylære bindinger – altså kræfter mellem forskellige molekyler. Lægemidler designes til at binde specifikt til enzymer eller receptorer og kan fungere som enten agonister, antagonister eller inhibitorer. Antagonister hæmmer receptorer, mens inhibitorer hæmmer enzymer eller proteiner. Et andet centralt aspekt i lægemiddeldesign er isomeri. Du kan læse mere om disse emner – samt om thalidomid-skandalen, hvor man overså betydningen af isomeri – i de følgende dropdowns.
Kendskabet til den rumlige opbygning og de kemiske egenskaber af proteinlommen på enten receptoren eller enzymet har stor betydning, når man skal udvikle et lægemiddel. Som eksempel kan lægemidlet ses som en nøgle, der skal passe i proteinlåsen for at lægemidlet virker (se figur 8).

Figur 8. Lægemiddel der vekselvirker med et aktivt center på et biologisk target.
Lægemidler binder som regel i en lomme på proteinets overflade. Bindingen kan både være covalent og ikke-covalent. Ved ikke-covalente bindinger binder lægemidlet gennem svage intermolekylære kræfter. Der findes flere typer intermolekylære kræfter, som normalt kan opstå mellem lægemiddel og target. Disse er nævnt nedenfor efter styrke, hvor ion-dipolbindinger generelt er de stærkeste, mens londonbindinger er de svageste:
- Ion-dipolbinding: Stærke kræfter mellem en ion og en polært binding. Ionen tiltrækkes af den modsatte ladning af molekylets dipol.
- Hydrogenbinding: Bindingen mellem et hydrogenatom og oxygen (O), nitrogen (N) eller fluor (F) i ét molekyle til oxygen (O), nitrogen (N) eller fluor (F) i et andet molekyle. Bindingen kan f.eks. opstå mellem en NH-gruppe og en C=O-gruppe.
- Dipol-pipol binding: Tiltrækning mellem to polære molekyler, hvor de positive og negative ender vender mod hinanden.
- London binding (Van der Waals kræfter): Dette er en hydrofob vekselvirkning. Svage tiltrækningskræfter, der opstår midlertidigt mellem alle atomer og molekyler på grund af bevægelige elektroner. De er de eneste kræfter i ikke-polære molekyler.

Figur 9. Her illustreres de fire forskellige former for intermolekylære kræfter.
Lægemidlets mange bindinger til target gør den samlede binding mellem lægemidlet og target stærkt. Hvis target f.eks. er et enzym, findes der flere måder, hvorpå lægemidler kan hæmme de forskellige processer i cellen. Et stof, som kan hæmme en proces, kaldes en inhibitor. Lægemidler kan enten være reversible inhibitorer (ibuprofen) eller ikke-reversible inhibitorer (acetylsalicylsyre). Ikke-reversible inhibitorer forårsager kemiske ændringer og ødelægger target ved at binde sig covalent til target. Reversible inhibitorer danner mange svage bindinger til target, men forårsager ingen kemisk ændring. Bindingen er derfor reversibel – heraf navnet. Reversible inhibitorer bruges ofte som lægemidler, da de i langt mindre grad giver bivirkninger.
En inhibitor kan yderligere opdeles i tre kategorier: kompetitiv, non-kompetitiv (også kaldet ikke-kompetitiv) og ukompetitiv. Ved kompetitiv hæmning kan inhibitoren binde i det aktive center og kæmpe om pladsen med substratet. En kompetitiv inhibitor er reversibel og kan udkonkurreres ved tilførsel af mere substrat. En non-kompetitiv inhibitor binder til et andet sted på enzymet (allosterisk site), ændrer strukturen i det aktive center og forhindrer dermed substratet i at binde sig. En ukompetitiv inhibitor binder til enzym-substrat-kompleks, hvorved reaktionen blokeres og ikke kan fuldføres. En non-kompetitiv og ukompetitiv inhibitor kan ikke udkonkurreres af øget mængder substrat, men de er stadig reversible inhibitorer og kan fjernes ved f.eks. nedbrydning, temperatur- eller pH-ændringer.
Som tidligere omtalt, har lægemidlers rumlige opbygning stor betydning for deres effekt i kroppen. Det skyldes, at lægemidler kan være chirale. Chirale molekyler har samme funktionelle sidegrupper, men er spejlbilleder af hinanden, og kaldes spejlbilledisomere (se figur 10). Det bedste eksempel på spejlbilledisomere er vores hænder. Lige meget, hvordan man vender og drejer dem, vil de altid være spejlbilleder af hinanden selvom begge hænder har både tommel-, pege-, lange-, ring- og lillefinger.

Figur 10. Den rumlige opbygning af carbonmolekylets sidegrupper kan optræde i forskellige former. De to molekyler er spejlbilleder af hinanden og kaldes spejlbilledisomere.
Et molekyle kaldes chiralt, når det indeholder et carbonatom, der er bundet til fire forskellige grupper. Dette carbon kaldes et chiralt carbonatom. For at finde ud af, hvilken form molekylet har, giver man de fire grupper en prioritet, alt efter hvilket grundstof de starter med. Gruppen med det højeste atomnummer i det periodiske system får højest prioritet, og den med det laveste får lavest prioritet. Når man har givet alle grupper en prioritet, vender man den lavest prioriterede gruppe væk fra sig selv, så man ser direkte på det centrale carbon og de tre andre grupper rundt om det. Hvis man derefter følger rækkefølgen fra højest til lavest prioritet og den går mod venstre, har molekylet S-form (S kommer af det latinske ord sinister, som betyder venstre). Går rækkefølgen i stedet mod højre, har molekylet R-form (fra rectus, som betyder højre på latin).
Et chiralt carbonatom kaldes også et stereocenter, og hvis et molekylet indeholder flere stereocentre, kan molekylet findes som flere stereoisomere. Stereoisomere kan enten være spejlbilleder af hinanden, hvor alle chirale centre er spejlbilleder, hvilket kaldes enantiomere. Eller kun nogle chirale centre er spejlbilleder af hinanden, hvilket kaldes diastereomere (se figur 11).

Figur 11. På dette billede ses fire stereoisomerer af aminosyren threonin. Stereocentrerne er angivet med en rød stjerne.
De fleste stoffer, der oprenses fra naturen eller fremstilles kunstigt i et laboratorium, har stereocentre. Det har vist sig, at det tit kun er en stereoisomer, som har den ønskede medicinske effekt. To forskellige stereoisomerer kan pga. deres forskellige strukturer virke på to fuldstændigt urelaterede receptorer, som hver især igangsætter to vidt forskellige biologiske processer (se dropdown om thalidomid-skandalen). Årsagen til dette er som omtalt orienteringen af de funktionelle sidegrupper (se figur 12).

Figur 12. Stoffet til venstre passer i receptoren, mens stoffet til højre ikke gør.
Betydningen af stereokemiske forhold i forbindelse med udviklingen af nye lægemidler har ført til, at der forskes meget indenfor fremstillingen af den rigtige stereoisomer med den ønskede medicinske virkning. I 2001 gik Nobelprisen i kemi til William Knowles og Ryoji Noyori for deres arbejde med fremstilling af stereoselektive kemiske synteser. W. Knowles fremstillede katalysatoren RhDiAMP, som bruges til syntesen af den ønskede stereoisomer af lægemidlet DOPA, som bruges til behandling af Parkinsons sygdom. Derved vil kun den ønskede stereoisomer af lægemidlet fremstilles.
Stoffet R-thalidomid lindrer morgenkvalme hos gravide kvinder, mens den anden stereoisomer forårsager alvorlige defekter på ufødte fostre, som f.eks. manglende eller misdannede organer eller lemmer.

Figur 13:Til venstre ses R-thalidomid, som er et sovemiddel, og til højre ses S-thalidomid, som er et teratogen.
I 1956 blev Thalidomid lanceret til behandling af morgenkvalme og søvnbesvær hos gravide kvinder. I starten af 1960’erne blev der indrapporteret en lang række svære fødselsdefekter hos nyfødte som bl.a. manglende arme og ben. Det førte til, at den tyske producent Grünenthal i 1961 måtte fjerne lægemidlet fra hylderne. Skandalen skabte røre over hele verden og i 1968 indførtes et rapporteringssystem for bivirkninger i Danmark. Selvom skandalen ikke var årsag til opdagelsen af stereoisomere, havde den en væsentlig betydning for forståelsen af og opmærksomheden på stereoisomerers biologiske betydning i medicinalindustrien.
Lægemidlerne skal ramme deres molekylære mål i hjernen
Ud over at skulle binde til et target i hjernen, skal lægemidlet også passere en lang række forhindringer i kroppen, før det når sit mål. Der stilles derfor store krav til stoffets biologiske og kemiske egenskaber. Inden for farmakologien hentydes der ofte til stoffets ADME-egenskaber. Et aktivt stofs ADME-egenskaber udtrykker, hvor godt stoffet absorberes, distribueres, omsættes og eliminering fra kroppen som de engelske navne også antyder (Absorption, Distribution, Metabolism, Excretion; ADME).
Absorption
De fleste lægemidler indtages oralt i form af piller. Det er derfor vigtigt, at det aktive stof i lægemidlet kan klare de kemiske forhold undervejs til target (som eksempelvis den lave pH i mavesækken). Stoffet skal kunne passere gennem tarmens cellemembran for at blive optaget i blodbanen. I nogle tilfælde er det dog ikke muligt, og det er derfor nødvendigt at indgive stoffet anderledes, f.eks. ved indsprøjtning eller inhalering. Lægemidler rettet mod det centrale nervesystem (CNS) skal desuden kunne krydse blod-hjerne-barrieren.

Figur 14. Illustration af kroppens naturlige barrierer. På figuren ses både mave-tarm-barrieren og blod-hjerne-barrieren.
I 1997 publicerede Christopher A. Lipinski en række egenskaber, som gennem hans arbejde havde vist sig at være vigtige for den orale biotilgængelighed af et givent lægemiddel. Disse regler kaldes Lipinski’s Rule of Five, da reglerne indeholder mange femtaller. For at opnå bedst mulig optagelse af et givent stof, skal det have følgende egenskaber.
- Molarmasse skal være mindre end 500 g/mol.
- Stoffets lipofilicitetsfordelingskoefficent skal være mindre end 5. Log P < 5.
- Antallet af hydrogenbindingsdonorer skal være mindre end 5.
- Antallet af hydrogenbindingsacceptorer skal være mindre end 10.
Morfin er et eksempel på et stof, som opfylder alle Lipinskis regler, men har moderat biotilgængelighed. Reglerne er ingen garanti for, hvorvidt et stof kan være en god lægemiddelkandidat. Et stof der ikke opfylder alle reglerne, kan sagtens have en tilfredsstillende biotilgængelighed. F.eks. hvis stoffet er meget potent, og det kun kræves i meget små koncentrationer for at have sin medicinske effekt.

| Formel: | C17H19NO3 |
| Molarmasse (g/mol): | 285,34 |
| Log P: | 1,27 |
| H-donerer: | 2 |
| H-acceptorer: | 4 |
Figur 15. Strukturformel og kemiskeoplysninger for morfin.
Distribution
Stoffer, som optages gennem epitelcellerne i tarmen, bevæger sig hurtigt over i blodbanen. Det er dog ikke alle molekyler, som opløses i blodet. Upolære molekyler opløses ikke, men transporteres af forskellige transportproteiner. Fra blodbanen distribueres stoffet til leveren og derfra ud til de forskellige væv og organer. Et stof siges at være effektivt, hvis det rammer målorganet i tilstrækkelig koncentration. Visse dele af kroppen er dog særlig svære at nå. Dette er f.eks. tilfældet med centralnervesystemet (CNS). På trods af den gode blodgennemstrømning i hjernen, er den svær at nå med aktive lægemidler pga. blod-hjerne-barrieren (BBB for engelsk blood-brain-barrier). BBB er opbygget af endotelceller, som sidder meget tæt for at forhindre passage af stoffer mellem cellerne. Derved opretholder BBB et konstant ekstracellulært miljø i hjernen. Upolære substanser diffunderer over BBB forholdsvis uhindret, mens polære molekyler skal transporteres af specielle transportproteiner. Dog kan polære molekyler nemmere opløses i blodet.

Figur 16. Strukturformlen af serotonin.
Neurotransmitteren serotonin har stor betydning for menneskers sindsstemning. Det har vist sig, at mange depressive har for lidt serotonin i hjernen. Serotonin er et eksempel på et stof, som ikke kan krydse BBB. Depression behandles derfor med alternative stoffer, som godt kan krydse BBB og dermed øge mængden af serotonin i hjernen. Disse stoffer kaldes SSRI-præparater.
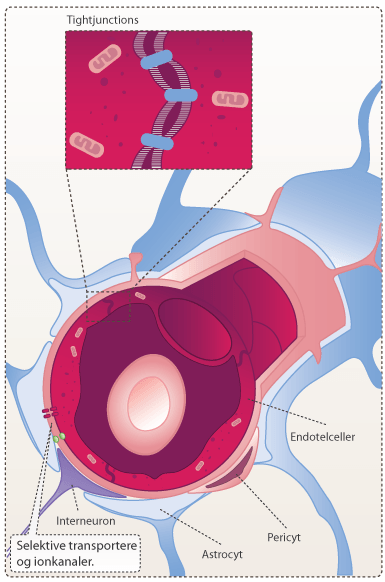
Figur 17. Blod-hjerne-barrieren (BBB). Barrieren har forskellige ionkanaler og transportere, som kan transportere aminosyrer, glucose og ioner over barrieren. Mange andre stoffer udelukkes dog.
Stofomsætning (metabolisme) og eliminering
De fleste stoffer nedbrydes i leveren af forskellige enzymsystemer, hvilket sker med forskellige hastigheder for de forskellige stoffer. Der findes stoffer, som i mindre grad end andre nedbrydes i leveren. Disse stoffer kaldes metabolisk stabile stoffer. Metabolismen af stoffer i leveren er en afgørende faktor, når nye lægemiddelkandidater skal udvælges, da nedbrydningshastigheden har stor betydning for stoffets effektivitet i kroppen. Stoffer, som omsættes for hurtigt i kroppen, egner sig ikke særlig godt som lægemidler, da de måske ikke når deres target i tilstrækkelig høj koncentration. Stoffer, der nedbrydes langsomt, kan ophobes, hvilket virker toksisk for kroppen.
Til sidst skal stoffet eller dets nedbrudte rester elimineres fra kroppen, hvilket kan ske gennem urin, afføring, sved, udånding med mere.
Test af lægemidler
Når forskere har identificeret en potentiel lægemiddelkandidat, påbegyndes en lang og grundig proces med at teste stoffet, både biologisk og kemisk. Indledningsvist udføres in vitro-forsøg, hvor stoffet undersøges uden for levende organismer – typisk i cellekulturer og med avancerede kemiske assays. Her forsøger man at besvare centrale spørgsmål som:
Hvordan trænger molekylet gennem biologiske membraner? Hvor fedtopløseligt og kemisk stabilt er det? Hvordan bliver det nedbrudt og udskilt fra kroppen? Og vigtigst: Er det giftigt?
Disse tidlige tests danner grundlaget for videre udvikling. De viser ofte, at det oprindelige molekyle skal ændres flere gange, før det har de rette egenskaber til at gå videre til test i levende organismer. De biologiske analyser in vitro foretages typisk i cellekulturer, og nogle af de mest anvendte celletyper beskrives i den første dropdown nedenfor.
Herefter følger en række prækliniske tests in vivo, hvor stoffets effekt og sikkerhed undersøges i forsøgsdyr – dette uddybes i anden dropdown. Til sidst, hvis resultaterne er lovende, kan lægemiddelkandidaten blive godkendt til test i mennesker, hvilket sker i fire veldefinerede kliniske faser. Disse gennemgås i den tredje dropdown.
HeLa kræftcellerne bruges i dag i en lang række biomedicinske sammenhænge bl.a. for at undersøge bindingen mellem et potentielt lægemiddel og dets biologiske target. Cellerne stammer oprindeligt fra den 31-årige Henrietta Lacks, som i 1951 blev indlagt på Johns Hopkins Hospital med en cancer tumor. På trods af at hun døde få måneder efter, lever hendes tumorceller stadig i dag, hvor de bruges i mange forskellige biologiske assays. HeLa kræftceller omtales tit som udødelige, da de kan dele sig uendeligt mange gange.

På Lundbeck bruger man både HeLa-kræftceller og kinesiske hamster-ovarieceller i forskningen. Cellerne gensplejses med moderne bioteknologi, hvor forskerne indsætter DNA, som koder for helt bestemte biologiske targets. Disse celler egner sig særligt godt til cellebaserede assays, hvor man f.eks. undersøger, hvordan et stof binder til eller påvirker en bestemt receptor eller et enzym. Både kinesiske hamster-ovarieceller og HeLa-kræftceller er velegnede til denne type analyser, da de deler sig hyppigt og er lette at gensplejse. I nogle assays bruger forskerne også rekombinant HIV-virus til at integrere udvalgte gensekvenser i cellekulturerne, da virussen er særligt effektiv til at indsætte DNA i cellens arvemateriale.
Efter en lang række in silico og in vitro tests er det tid til, at teste lægemiddelkandidaterne på levende organismer in vivo. Inden lægemidlet gives til mennesker, gennemtestes kandidatens effektivitet og toksikologi på forsøgsdyr. Der bruges mange forskellige slags forsøgsdyr, dog oftest mus og rotter. I Danmark kræver det godkendelse for at arbejde med forsøgsdyr. De enkelte forsøg skal også godkendes af Dyreforsøgstilsynet, som hører under Justitsministeriet.
En af metoderne, der kan bruges til at undersøge et lægemiddels indvirkning på hukommelse og indlæring, er forsøg i Morris water maze (Morris vandlabyrint). Morris water maze ville kunne være relevant til test af lægemidler mod sygdomme som f.eks. Alzheimers sygdom og skizofreni, hvor der ses tab af hukommelse og indlæringsevne. I forsøget sættes en rotte i en stor vandtank fyldt med mælket vand. Under overfladen i den ene del af karret er der en lille platform, som giver rotten mulighed for at undgå at svømme. En normal rotte vil svømme rundt, indtil den finder platformen. Anden gang rotten bliver placeret i bassinet, vil den svømme direkte hen til platformen. I det ikke-normale tilfælde, som f.eks. ved hukommelsessvigt ved Alzheimers eller skizofreni, vil rotten have svært ved at huske placeringen af platformen og derfor svømme rundt uden at finde den hurtigt anden gang.

Figur 19. PCP blokerer ionkanalen i NMDA-receptoren.
Ved at give rotten stoffet PCP, som blokerer NMDA-receptoren, svækkes rottens indlæringsevne og andre kognitive funktioner, hvilket også er karakteristisk for personer med skizofreni. Lægemidler mod Alzheimers eller skizofreni kan nu testes på rotter, som har fået PCP. Hvis lægemidlet får rotten til hurtigt at finde platformen i anden omgang, har lægemidlet virket. Morris water maze er en ud af flere metoder, som forskerne kan bruge til at analysere og tolke dyrenes adfærd. Disse metoder hjælper forskerne til at forstå, hvordan lægemiddelkandidaterne virker, inden de testes i mennesker.

Figur 20. Morris vandlabyrint. En normal rotte vil hurtigt lære, hvor platformen befinder sig i tanken, og derfor vil den have let ved at finde den anden gang, forsøget udføres. Den PCP-påvirkede rotte vil bliver ved med at have problemer med at finde platformen, også anden, tredje og fjerde gang forsøget udføres. Dette skyldes PCPs påvirkning af rottes indlæring. (Kør musen henover billedet og se forskellen på de to rotters adfærd).
Når nye lægemidler udvikles, skal de naturligvis også testes i mennesker. I Danmark kræver myndighederne, at nye lægemiddelkandidater først er gennemtestet for toksikologi og bivirkninger i forsøgsdyr. Herefter kan lægemiddelkandidater blive godkendt af Lægemiddelstyrelsen og Den Centrale Videnskabsetiske Komité til at gå over i de fire kliniske faser. De kliniske faser er langt den dyreste del i udviklingen af nye lægemidler. Ud af f.eks. 5.000-10.000 stoffer er der måske kun fem, som går videre til de kliniske faser, og heraf kun et stof, som bliver et lægemiddel.
- Fase 1 (Sikkerhed): I første fase gives lægemidlet til en lille gruppe af raske frivillige (10 til 100 personer), og der igangsættes en undersøgelse af, hvordan den menneskelige organisme optager og reagerer på stoffet. I løbet af forsøget modtager forsøgspersonerne flere forskellige doser af stoffet. Dog meget mindre doser end en normal dosis, idet der holdes skarpt øje med tegn på forgiftning. I denne fase fokuseres der primært på, om lægemidlet har en giftig virkning i mennesket. Effektiviteten af stoffet evalueres ikke nødvendigvis.
- Fase 2 (Sikkerhed, effektivitet, dosering): I anden fase gives lægemidlet til en lidt større gruppe af frivillige (50 til 300 personer), som vil kunne have gavn af lægemidlet. Gennem denne fase undersøges forskellige doser for deres effektivitet og bivirkninger. Som regel udføres der sideløbende kontrolforsøg, hvor kontrolgruppen modtager placebo i stedet for det rigtige lægemiddel. Hverken forskere eller forsøgspersoner ved, hvilke grupper, der får hvad. Til sidst i forsøget sammenlignes doser og bivirkninger fra de to grupper, og de mest optimale doser bestemmes.
- Fase 3 (Sikkerhed, effektivitet, bivirkninger): I tredje og sidste fase udføres forsøg i stort omfang med mange frivillige (1500-4000 personer), som vil kunne have gavn af lægemidlet. Lægemidlet gives i de doser, som man tidligere har fundet passende. I løbet af forsøget går forsøgspersonerne til kontrol hos deres egen læge, hvor de rapporterer om bivirkninger. Denne undersøgelse giver mulighed for at se, om lægemidlet giver sjældne bivirkninger, dvs. bivirkninger hos nogle få af forsøgspersonerne.
- Fase 4: I denne fase er lægemidlet godkendt af myndighederne og vil nu kunne bruges til behandling af sygdommen. I denne fase laves der tit forsøg, hvor lægemidlet sammenlignes med allerede eksisterende behandlinger.

Figur 21. Oversigt over faserne i lægemiddeltest.
De meget omfattende undersøgelser, som skal udføres gennem de kliniske faser, gør det meget kostbart at udvikle lægemidler. Udviklingen af et nyt lægemiddel er estimeret til at koste omkring 800 mio. dollars. Selv om et lægemiddel kommer på gaden, kan det stadig blive taget af hylderne, hvis det ikke fungerer efter hensigten. Ligesom det var tilfældet med thalidomid. Efter endelig godkendelse og etablering på markedet holdes der altså stadig øje med nyopståede bivirkninger eller toksiske effekter.